
What Is Hypertrophic Cardiomyopathy?
Hypertrophic cardiomyopathy (HCM) affects one in 500 people in the general population. In most cases, HCM is caused by genetic mutations.
Doctors usually discover HCM during cardiac testing (an electrocardiogram or echocardiogram). During this test, doctors see thickening (hypertrophy) on the heart’s left lower chamber (ventricle), even though the patient has no other disease that could cause this thickening.
Most patients with left ventricular hypertrophy have heart cells that are larger than normal. These larger cells cause the heart's lower chambers, usually the left chamber (ventricle), to become thick and stiff. But the location and amount of thickening can be very different for each person. Because of this, HCM can be hard to diagnose.
Why Choose University of Utah Health?
Comprehensive, Expert Care for Treating HCM

The Hypertrophic Cardiomyopathy (HCM) Program at University of Utah Health is the first in the state of Utah and one of the nation’s few comprehensive programs for evaluating and managing HCM. The program offers a comprehensive evaluation by experts in:
- cardiomyopathy,
- cardiac imaging,
- cardiac genetics,
- arrhythmia,
- interventional cardiology, and
- cardiac surgery.
We understand that hypertrophic cardiomyopathy is a complex disease. By bringing doctors from multiple specialties together, we can provide leading-edge evaluation and a therapeutic plan that best suits your individual needs.
Find a Hypertrophic Cardiomyopathy Specialist
How Is Hypertrophic Cardiomyopathy Diagnosed?
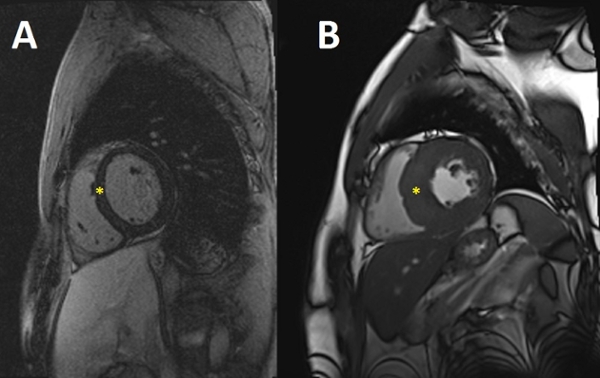
Figure A shows a normal patient's
heart; figure B shows a patient's
heart with hypertrophic
cardiomyopathy.
Doctors often diagnose HCM by using noninvasive cardiac imaging, including echocardiography and/or cardiac magnetic resonance imaging. To diagnose HCM, doctors will look at a patient's left ventricle (lower heart chamber). If the patient's left ventricle is hypertrophied or thickened (usually >=15 mm wall thickness), and if the patient doesn't have cardiac disease or another disease that causes ventricle thickening, the patient may have HCM.
Inherited Disorder
HCM is inherited in an autosomal dominant pattern. This means that people with HCM have a 50 percent chance of passing this condition on to their children. Genetic researchers have identified over 1,500 mutations affecting more than 15 genes associated with HCM.
HCM can also be identified through family history and molecular genetic testing. These tests accurately diagnose HCM in a patient. They can also identify if a patient's relatives have the HCM mutation.
You can have genetic testing for HCM with a blood sample. The test is covered by most insurance policies.
HCM Treatment Options
Treatments We Provide



Hear From Our Specialists

For more information email us at hcm@hsc.utah.edu. Call 801-585-5122 or
Request an Appointment